Chip microarrays
ChIP- chip microarrays to study the epigenome in leukemia
Authors: C. Müller-Tidow and M. Dugas, Department of Medicine A, Hematology and Oncology and Department of Medical Informatics and Biomathematics, University of Münster; Germany Information Letter No.5, August 2008
Microarrays to study leukemia
High throughput analyses have revolutionised research approaches in molecular biomedicine. The best known and established approach, the global analyses of mRNA-expression by microarrays, is now commonly used in leukemia research. Profiling of leukemia mRNA can also be used for diagnoses and classification. Recent developments in microarray technology and specimen-preparation have also led to the development of novel and complementary techniques. It is now possible to identify direct genomic binding sites of transcription factors as well as genome wide chromatin modifications by microarrays 1, 2. This is of special importance for leukemia, since transcription factor alterations are of crucial importance in the pathogenesis of several leukemia subtypes. For example, balanced translocations in acute myeloid leukemia (AML) involve transcription factors that are commonly associated with proliferation and / or myeloid differentiation.
Identification of their direct genomic targets as well as the identification of genome-wide epigenetic alterations in AML might lead to better understanding of the disease and to novel therapeutic targets.
Principle of ChIP-Chip
These novel microarray applications rely on different array platforms and usually originate from the specific immunoprecipitation of a protein-DNA complex (Fig. 1). They are thus called chromatin- IP-coupled with microarrays (ChIP-Chip analysis). First, proteins and DNA are cross-linked by formaldehyde in living cells in vivo. Subsequently, chromatin is sheared into smaller fragments with the transcription factors and the chromatin modifications still being in place. Sensitive and specific antibodies are utilized to significantly enrich specific protein-DNA-complexes from the total pool of chromatin. The immunoprecipitated DNA is purified. Finally, the DNA is amplified, labelled and hybridised ontogenomic microarrays. The fluorescence intensity of the different genomic locations contained on the microarray can then be analysed and genomic binding sites can be correctly determined by appropriate bioinformatics analyses. In contrast to microarrays for expression profiling, genomic arrays most importantly contain promoter and enhancer as well as locus control regions, which are relevant for transcription factor binding and regulation of gene expression. There are already several sources for specific genomic oligoarrays. Besides a few commercial suppliers, we have established a novel microarray-platform based on 50 mer oligonucleotides. These more than 33,000 independent enomic oligonucleotides are placed in the promoters and regulatory regions of more than 11,000 well characterised protein coding genes as well as more than 500 miRNAs. This microarray platform is very versatile given that novel sequences can easily be added. This is important since 2nd generation sequencing methods can be used after chromatin immunoprecipitation to identify transcription factor binding sites or chromatin alterations on a genome wide scale in vivo. The resulting spots (for example discovered in few samples in cell-lines) can then be used to complement the genomic oligoarray. Finally, the custom array can be used to query patient specimen with high throughput at relatively low cost.
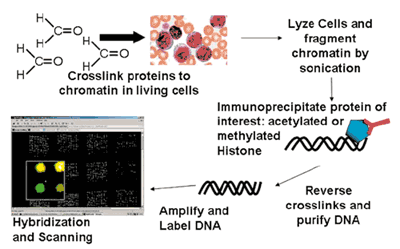
Fig. 1: Principle of ChIp-Chip Arrays. ChIP-Chip assays are used for microarray based analysis of global histone modification patterns (shown in Figure) or for transcription factor targets in vivo. Chromatin complexes containing DNA and protein are immunoprecipitated using specific antibodies. Subsequently, the DNA immunoprecipitated along the protein is purified and finally hybridized onto genomic microarrays.
Applications for ChIP-Chip and genomic arrays
Regardless of the choice of array platform or even the use of 2nd generation sequencing to analyse the chromatin IPs 3, three major arrays of application are emerging. First, transcription factor binding can be profiled with high sensitivity and specificity. Second, chromatin modifications such as posttranslational modifications of histones can be analysed. Histone modifications such as acetylation or methylation of amino acid residues are crucially important not only for chromatin structure but also for transcriptional regulation. Multiple histone modifications have been discovered and these are indicators of promoter activity and act as docking molecules for signalling. Finally, DNA-methylation can be analysed by immunoprecipitation of methylated DNA using antimethylated- Cytosine antibodies. An alternative for the latter approach is the use of methyl-CpG binding proteins to identify methylated DNA-regions. Thus, genomic arrays can be used for a wide spectrum of in vivo analyses regarding transcriptional regulation and genome organization.
Applications of ChIP-Chip in leukemia research
We have recently used ChIP-Chip to identify genomic binding sites of PMLRARα, the chimeric oncogene produced by t(15;17) in acute promyelocytic leukemia (Fig. 2). More than 300 direct genomic in vivo targets of PML-RARα could be identified 4. This number far exceeds the only handful of already known direct PML-RARα targets. The PML-RARα targets can be broadly placed in genes regulating apoptosis, proliferation, transcription and myeloid differentiation. This is very well in accordance with the known biological functions of PMLRARα. In addition to identifying PMLRARα target genes, we also analysed the biological consequences of genomic PML-RARα binding. We utilised the same ChIP-Chip-platform to analyse the binding of histone deacetylase 1 as well as the alteration of several histone modifications in PML-RARα expressing versus non expressing cells. Interestingly, these data showed that PML-RARα acts as universal repressor on almost all of its presumed direct targets. These findings highlight the special power of ChIP-Chip analyses for the identification of transcription factor targets in vivo. Recently, we have started to identify global alterations of important histone modifications in primary AML patients. In this currently ongoing project there are more than 170 patients with acute leukemia and control patients being analysed by ChIP-Chip for global histone acetylation and histone methylation pattern. Importantly, these analyses led to the generation of histone modification signatures that unambiguously allowed the identification of AML specimen versus normal hematopoietic progenitors and also allowed to distinguish between AML and ALL . In each case, between several hundreds and thousands of altered genomic locations could be identified by ChIP-Chip analyses. With the resulting data sets we can now analyse similarities and differences between individual leukemia subtypes and we are able to look for larger regions of epigenetic deregulation in vivo.
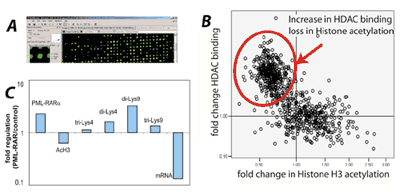
Fig. 2: ChIP-Chip analysis to identify PML-RARα target genes.
A) We analyzed genome wide binding of PML-RARα using ChIP-Chip. Example of an array.
B) Identified PML-RARα target genes were analyzed for the resulting chromatin modifications. Each target gene is represented by one dot. Many target genes showed increased binding of histone deacetylase 1 (HDAC) and subsequent loss of histone acetylation.
C) The PML-RARα target genes (here one example) showed a repressed chromatin configuration (increase in methylation of lysine 9 di-Lys9, tri-Lys 9 and loss of acetylated H3-acH3). As a consequence, mRNA expression is repressed.
Outlook
ChIP-Chip analyses have recently emerged as powerful tools for cell-culture-based analyses. Recent data from our laboratory also show that ChIP-Chip analyses can be successfully employed for the analyses of primary patient specimen. For in vitro as well as in vivo analyses, ChIP-Chip is increasingly used for the analysis of leukemia pathogenesis. Another prospect for the future could be to use ChIP-Chip for leukemia classification with the prospect of identifying so far unknown leukemia subtypes. In addition, the development of epigenetic drugs in AML, such as HDAC inhibitors and DNA methylation inhibitors, could be supported by ChIP-Chip analyses that identify response predictors in vivo. In the near future, the increasing availability of 2nd generation sequencing will complement ChIP-Chip analyses. These technologies will significantly increase our knowledge about leukemia pathogenesis in the near future and lead to novel therapeutic targets.
References
- Lee, T. I., Johnstone, S. E., and Young, R. A. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat Protoc, 1: 729-748, 2006.
- Ren, B., Robert, F., Wyrick, J. J., Aparicio, O., Jennings, E. G., Simon, I., Zeitlinger, J., Schreiber, J., Hannett, N., Kanin, E., Volkert, T. L., Wilson, C. J., Bell, S. P., and Young, R. A. Genome-wide location and function of DNA binding proteins. Science, 290: 2306-2309, 2000.
- Barski, A., Cuddapah, S., Cui, K., Roh, T. Y., Schones, D. E., Wang, Z., Wei, G., Chepelev, I., and Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell, 129: 823-837, 2007.
- Hoemme, C., Peerzada, A., Behre, G., Wang, Y., McClelland, M., Nieselt, K., Zschunke, M., Disselhoff, C., Agrawal, S., Isken, F., Tidow, N., Berdel, W. E., Serve, H., and Müller-Tidow, C. Chromatin modifications induced by PML-RARalpha repress critical targets in leukemogenesis as analyzed by ChIPChip. Blood, 111: 2887-2895, 2008.